R maaslin2 関数: オプションと実例
UB3/informatics/r/maaslin
このページの最終更新日: 2026/04/09関連ページ
広告
概要: Maaslin とは
Maaslin は Microbiome Multivariable Association with Linear Models の略で、腸内細菌にさまざまな 回帰分析 を適用し、腸内細菌の変動に影響する要因を解析するパッケージである。
GitHub ページ に情報がある。
インストールは Bioconductor から。BiocManager::install("Maaslin2") とする。単に install.packages("Maaslin2") ではダメだった (2023 年 7 月)。
help(Maaslin2) でヘルプを見ることができ、バージョン情報もここに含まれている。
PERMANOVA という手法を使って腸内細菌叢に影響する要因を大まかに特定し、Maaslin を使って詳しく解析するという手法がとられるらしい (1)。
広告
「あとがき」で当サイトを参考にしたと書いてくれているラノベです。Kindle Unlimited で読めました。ストーリーと文章が良く、面白かったです。
Maaslin のインプット
現在は Maaslin2 というパッケージが主に解析に使われている。script の基本はこんな感じ。
Maaslin2 には、2 つの input files が必要である (3)。
データファイル |
列に feature、行に sample を並べる。Microbes, genes, pathways など。transpose しても OK と書かれている。 |
メタデータファイル |
同様に、列に feature、行に sample が基本だが、transpose されていても OK。 |
2 つのファイルで sample が一致している必要はなく、どちらかで欠けている sample は自動で解析から除かれる。順番も違っていて大丈夫なようだ。
文献 3 で言及されている Maaslin2 source は、文献 4 の GitHub ページのこと。data および metadata の例として、HMP2_taxonomy.tsv と HMP2_metadata.tsv がそれぞれ挙げられている。一部をスクリーンショットで載せておこう。
データファイル HMP2_taxonomy.tsv はこういう感じ。ID があり、バクテリアの量が数値で表されている。
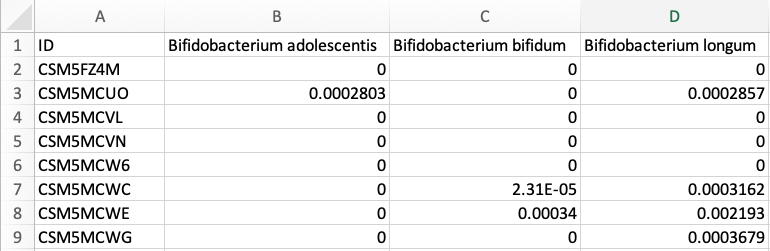
メタデータファイル HMP2_metadata.tsv は、同じ ID をもち、バクテリアの数値の代わりに、対象者の年齢、diagnosis などのデータが並ぶ。この 2 つのファイルを読み込み、変数として指定する。
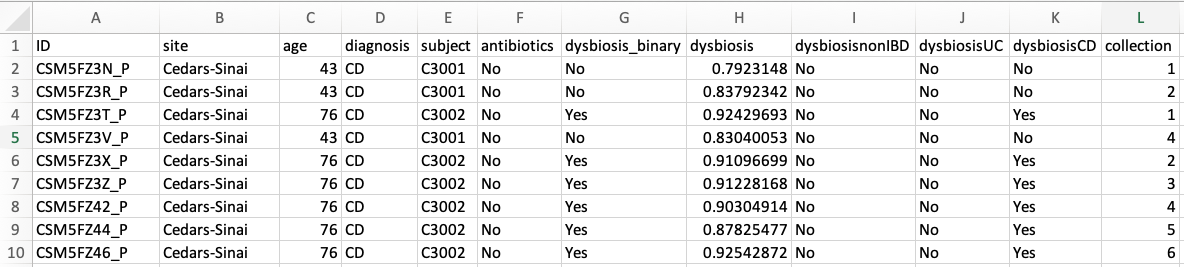
その他、指定するパラメーターについても表を作っておく。
"output_folder_name" |
" " の中に、アウトプットファイルを保存するフォルダの名前を指定する。あまり重要ではないだろう。 |
fixed_effects |
メタデータファイルの列の 1 つを "site" のような形で指定する。Maaslin は基本的には回帰分析なので、1 つを指定すると単回帰分析になる。 ここで、複数の列を指定することもできる。c("site", "age") のようにすると、複数の変数を指定できるようになる。 |
min_prevalence |
Maaslin2 の公式ページ によると、The minimum percent of samples for which a feature is detected at minimum abundance と定義されている。つまり、minimum abundance 以上で、特定の feature が検出されるサンプルの割合である。 このパーセントという言葉が紛らわしいが、通常は min_prevalence = 0.01 とした場合、それは 0.01% でなく、0.01 = 1% を意味する。 min_abundance = 0、min_prevalence = 0.01 の場合、マイクロマイオームでは「その菌が検出される (0 以上) の人が、1%以上存在する菌種を対象に maaslin 解析する」ということになる。 いずれ別のページに移すが、min_prevalende でカットオフする R script は以下の通り。 min_prev_cutoff <- 0.025 |
random_effects |
この扱いは難しい。このページ には、以下のようなことが書かれている。
その他の参考ページ: 固定効果とランダム効果について、Fitting problem only when using random effects、Confounding factors など。 |
reference |
'変数, reference' の形をとり、カテゴリー変数 categorical variable で reference として使う level を指定する。具体例は、ページ下の カテゴリー変数を説明変数に含める を参照のこと。 Categorical variable については、尺度水準のページ を参照のこと。 |
normalization |
デフォルトは TSS (おそらく total sum-scaling ) である。データが既にこの方法でノーマライズされているときは、 normalization = 'NONE' を指定する (参考)。 |
Maaslin のアウトプット
all_results.tsv と significant_results.tsv というファイルが出てくる。Official manual (Ref. 4) によると、all_results.tsv は全てのデータが q value の順に並べられており、ここから別途設定した q の閾値で有意なものだけが significant_results.tsv に含まれる。
閾値は -s MAX_SIGNIFICANCE で指定し、デフォルトの値は 0.25 である。
アウトプットでは、coef という値が出てくる。これは the model coefficient value で、 効果量 effect size のことであると書かれている (2,3)。しかし、文献 2, 3 のスレッドによると、
実際の解析例
では、HMP2_taxonomy.tsv と HMP2_metadata.tsv を実際に input し、解析を実行してみる。
まずは、データを読み込んで fixed_effects を連続変数である age にして解析。ID の列を、ページ後半の解析に合わせてちょっといじっている。
なお、2023 年 1 月にスクリプトの間違いを発見、修正した。前のスクリプトには %>% select(-ID) が含まれておらず、ID が菌の量を示したデータに含まれてしまっていた。
input_metadata の方は、fixed_effects で使う行を指定しているので、もとのファイルに何が含まれていても大丈夫である。しかし、結果を比べてみると、coefficient は ID が入っている場合も入っていない場合も完全に同一だった。log をみてみると、ID の列は解析に含まれていなかった。連続変数でないと、自動的に除くようになっているのかもしれない。
これを実行すると Example_output というフォルダができ、その中に結果のファイルが保存される。各ファイルの詳細については、ページ下方の Maaslin のアウトプット を参照のこと。ここでは、プロセスの理解を目的にメインの結果のみ載せていく。
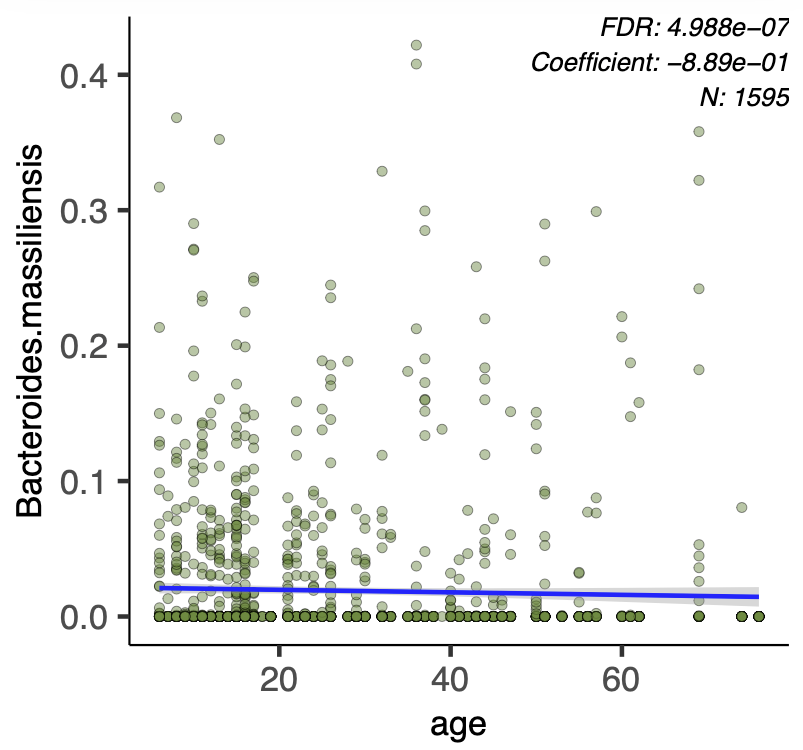
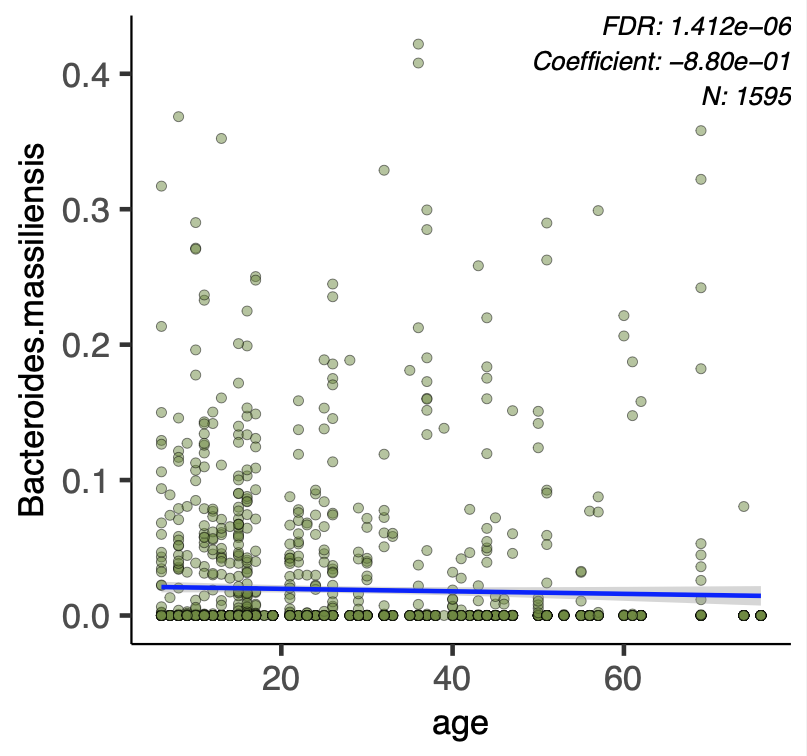
age.pdf という 59 ページの pdf ファイルができる。このファイルには、指定した要因と菌種のプロットがある。
一例を挙げると、この図。縦軸には input.data に含まれる菌の一つ、Bacterioides. massiliensis の abundance、横軸には age がプロットされており、回帰直線がある。Maaslin は 回帰分析 であることがよくわかる図である。
このような回帰分析が、複数の菌種に対して行われているので、P 値は FDR で補正されている。Coefficient は回帰係数のようなもので (これも Maaslin のアウトプット 参照)、この場合は弱い負の相関があるため、マイナスの値になっている。N は 1595。
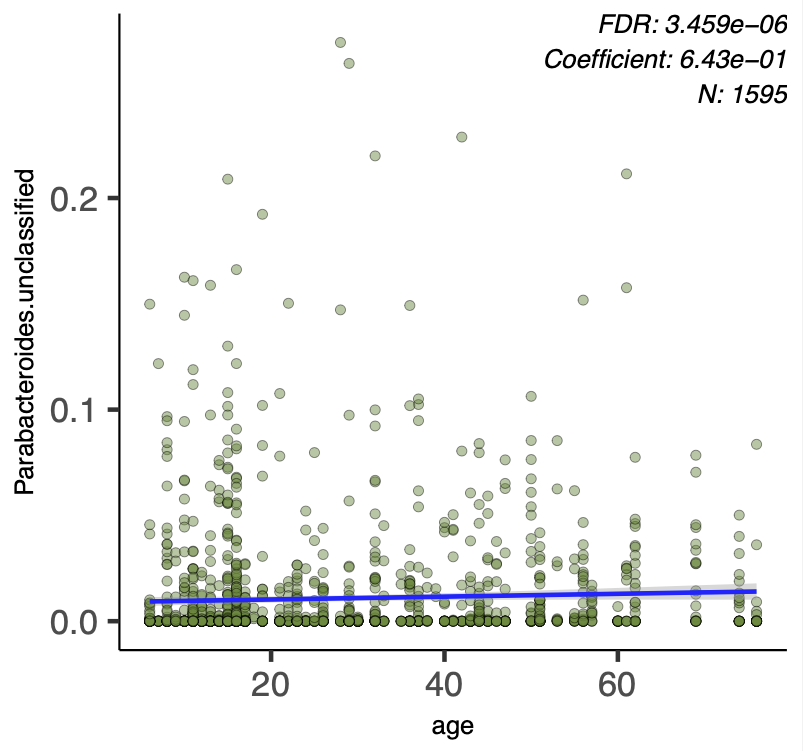
他の菌種では、弱い正の相関が認められ、coefficient は正の値である。
all_results.tsv というファイルには全てのデータが含まれる。input_data には 87 の菌種が含まれ、all_results.tsv ではそれらの全てについて coef や p val が計算されている。ヘッダーを含め 88 行のファイルである。
次に、要因を 2 つ指定してみる。
同じ菌種で、データの分布は同じ (はず) だが、coefficient と FDR が微妙に違っており、もう一つの要因である dysbiosis での調整が入っていることがわかる。
dysbiosis.pdf というファイルも作られる。同じような回帰分析のプロットが入っている。
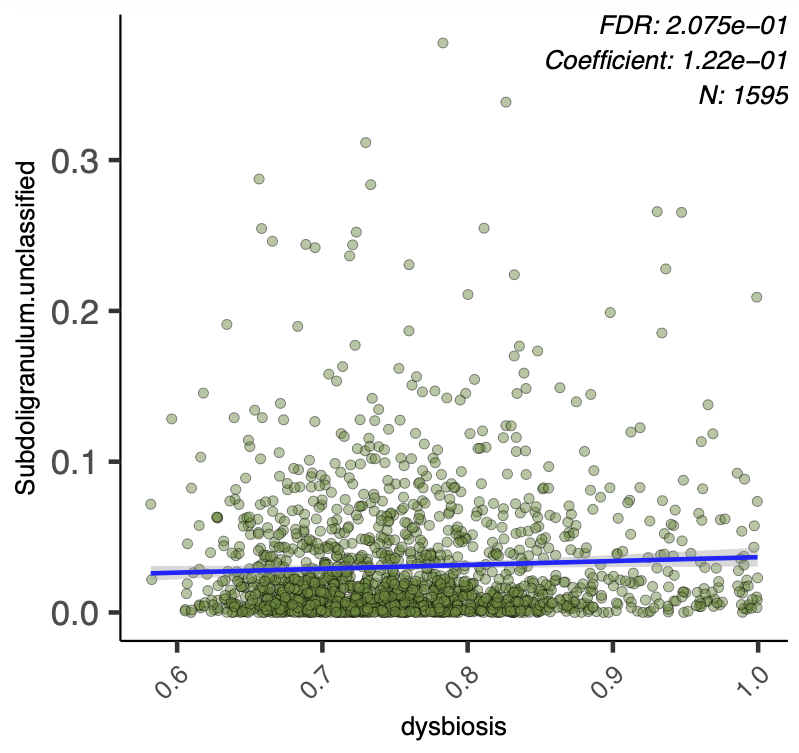
all_results.tsv は、87 菌種 x 2 要因 + ヘッダー行の 175 行のデータであり、そのうち有意なものだけが significant_results.tsv に含まれる。
さらに要因を追加。metadata で Yes or No のバイナリデータである antibiotics を要因に加えてみる。
二値変数なので、プロットは以下のようになる。従属変数が二値ならロジスティック回帰だが、この場合は説明変数が二値であり、線形回帰の範疇に入るようだ。
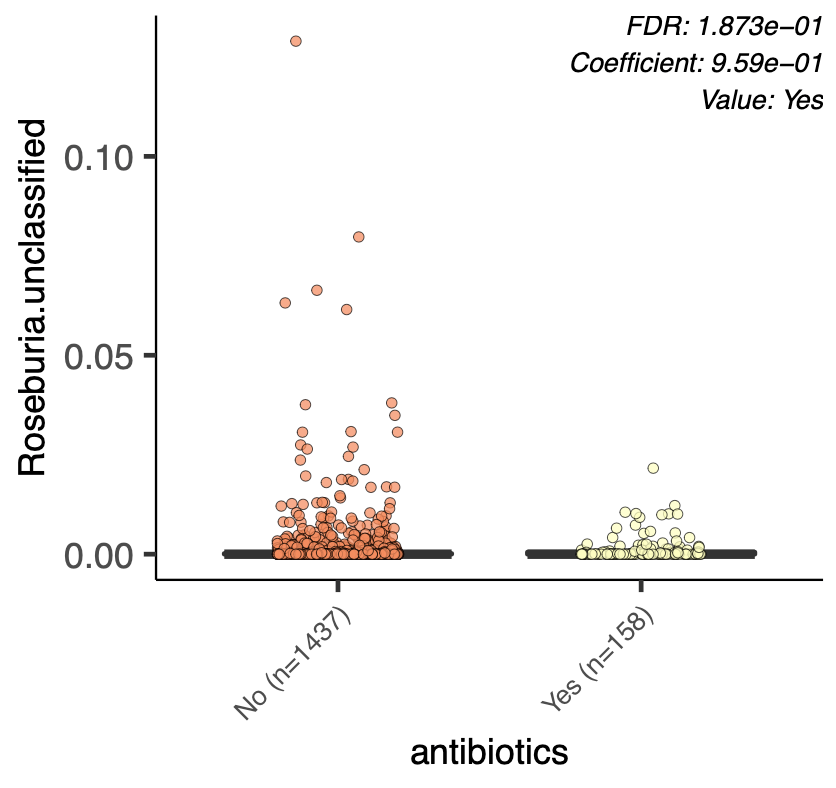
maaslin2.log というログファイルを見ると Formula for fixed effects: expr ~ age + dysbiosis + antibiotics という部分があり、モデルにどの因子が含まれているかを確認することができる (2)。
カテゴリー変数を説明変数に含める
メタデータで、diagnosis は CD, UD, nonIBD という 3 つの値を含むカテゴリー変数である。この場合、一番下の行のように reference となる変数を指定しなければならない。この行がないと Please provide the reference for the variable 'diagnosis' which includes more than 2 levels: CD, UC, nonIBD. というエラーになる。
Maaslin2 メモ
2022 年 5 月、xtfrm.data.frame(x) : cannot xtfrm data frames というエラーメッセージ。新しいバージョンの R に関係するエラーで、実際に問題を起こすことはないという ページ がある。
set.seed(1) のようにしても値は変わらないので、seed は使っていないようだ。
バージョン 1.14.1 アップデート時のエラー
2023 年 8 月、バージョン 1.14.1 にアップデートしてから遭遇したエラー。
まず、同じスクリプトを実行すると "If supplying input_data as a data frame, it must have appropriate rownames!" というエラーが。
行の名前がないとダメというエラーに見えたので、rownames() で名前をつけてみたが解決せず。このページ では、read.csv によるデータ読み込みに以下のオプションを追加して解決ししている。
これは、csv の 1 列目を行の名前にするということで、1 列目の内容によってはこれでエラーが解決する。ただし、どうもこの appropriate row name というのは、行名が数字だけだと appropriate と判断してくれないような気がする。数字だけの場合は、適当な文字を付け足して 1A とかにすると動くようになる (参考: ベクターの要素に文字列を追加する)。
なお、input_data と input_metadata の行名が異なっていると、"Unable to find samples in data and metadata files. Rows/columns do not match." というエラーでストップしてしまう。
広告
References
- MaAsLinの使い方のメモ. Link: Last access 2022/05/06.
- Coefficient in maaslin2 output. Link: Last access 2022/05/15.
- Maaslin2. Link: Last access 2022/05/15.
- GitHub Official manual. Link: Last access 2022/05/15.
広告
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。