Mac でベイズ法の系統樹を作る:
MrBayes のインストールと使い方
UB3/informatics/bioinformatics/tree_make_mrbayse_mac
広告
概要: MrBayes とは
2022 年 1 月ぐらいから本格的に使い始めた。それ以前の古い情報は、MrBayes 古い情報 のページへ移動。主に Mr. Bayes 3.2.7a について。系統樹に関する基本的な事項は、系統樹の基礎 のページなどを参照のこと。
インストールは homebrew で簡単にできた。BEAGLE なども dependency として自動でインストールされる。Ubuntu の場合は sudo apt-get install mrbayes で OK (2022 年 3 月、20.04)。
使い方の概要は以下の通り。
MrBayes の起動とデータ読み込み
ターミナル で起動、読み込みを行う。MrBayes を起動する前に、カレントディレクトリに nexus ファイルを準備しておく。
を実行すると起動され、プロンプトが MrBayes > に変化する。ここで以下のように execute を利用してファイルを読み込む。
読み込みむのは nexus 形式のファイルで、拡張子は nexus, nxs など。このファイルを作る方法はいくつかある。
- Clustal Omega のアウトプットを nexus にし、結果を右クリックでダウンロードすると clustalo-長い数字-.nxs というファイルがダウンロードされる。CIPRES ではこれをさらに変換しなければならなかったが、ローカルの MrBayes ではそのまま使える。
- T-coffee, M-coffee の結果を nexus にするときは、まず fasta フォーマットの結果をダウンロードする。これを EMBOSS seqret で nexus に変換できる。nexus/paup interleaved format と nexus/pauo non-interleaved があるので、interleaved の方を選ぶ。ただし、このページは DNA, protein の設定が正しく認識されない気がする。nexus ファイルをマニュアルで変更する必要があるかもしれない。datatype=DNA または Protein、missing=N または X とした。
- MUSCLE アラインメントは、この EMBL で output format 結果を ClustalW にする。これを上記の EMBOSS seqret で nexus に変換する。
モデルの決定
MbWiki には、以下の例がクイックスタートの方法として載っている (1)。
ここでは、2 つのパラメーターを設定している。lset では、likelihood model のパラメーターを設定する (2)。この例では、nst の 6 番 (general model of DNA substitution, the GTR) と、gamma-distributed rate variation accross sites に設定しているようだ。
選択できるモデルは多数ある。まずは、
- タンパク質をコードしていない塩基配列: MrModeltest と PAUP を使ってモデルを決定する。
- タンパク質をコードしている塩基配列: コドン内の位置によって異なる進化モデルを使うのが普通。 文献 5 では FindModel というウェブサイトを使っている。
- アミノ酸配列: Prottest モデルを決定する。
このページ にあった MrBayes の置換モデル一覧。
| Algorithm | Settings |
|---|---|
| GTR | lset applyto=() nst=6 |
| GTR + I | lset applyto=() nst=6 rates=propinv |
| GTR + G | lset applyto=() nst=6 rates=gamma |
| GTR + G + I | lset applyto=() nst=6 rates=invgamma |
| SYM + gamma | lset applyto=() nst=6 rates=gamma |
| prset applyto=() statefreqpr=fixed(equal) | |
| SYM + I + gamma | lset applyto=() nst=6 rates=invgamma |
| prset applyto=() statefreqpr=fixed(equal) | |
| SYM | lset applyto=() nst=6 |
| prset applyto=() statefreqpr=fixed(equal) | |
| SYM + I | lset applyto=() nst=6 rates=propinv |
| prset applyto=() statefreqpr=fixed(equal) | |
| SYM + gamma | lset applyto=() nst=6 rates=gamma |
| prset applyto=() statefreqpr=fixed(equal) | |
| SYM + I + gamma | lset applyto=() nst=6 rates=invgamma |
| prset applyto=() statefreqpr=fixed(equal) | |
| HKY | lset applyto=() nst=2 |
| HKY + I | lset applyto=() nst=2 rates=propinv |
| HKY + gamma | lset applyto=() nst=2 rates=gamma |
| HKY + I + gamma | lset applyto=() nst=2 rates=invgamma |
| K2P | lset applyto=() nst=2 |
| prset applyto=() statefreqpr=fixed(equal) | |
| K2P + I | lset applyto=() nst=2 rates=propinv |
| prset applyto=() statefreqpr=fixed(equal) | |
| K2P + gamma | lset applyto=() nst=2 rates=gamma |
| prset applyto=() statefreqpr=fixed(equal) | |
| K2P + I + gamma | lset applyto=() nst=2 rates=invgamma |
| prset applyto=() statefreqpr=fixed(equal) | |
| F81 | lset applyto=() nst=1 |
| F81 + I | lset applyto=() nst=1 rates=propinv |
| F81 + gamma | lset applyto=() nst=1 rates=gamma |
| F81 + I + gamma | lset applyto=() nst=1 rates=invgamma |
| Jukes Cantor | lset applyto=() nst=1 |
| prset applyto=() statefreqpr=fixed(equal) | |
| Jukes Cantor + I | lset applyto=() nst=1 rates=propinv |
| prset applyto=() statefreqpr=fixed(equal) | |
| Jukes Cantor + gamma | lset applyto=() nst=1 rates=gamma |
| prset applyto=() statefreqpr=fixed(equal) | |
| Jukes Cantor + I + gamma | lset applyto=() nst=1 rates=incgamma |
| prset applyto=() statefreqpr=fixed(equal) |
私が使ったことのあるモデルを中心にまとめておく。網羅できていないと思われるので、公式サイトも参照のこと。
WAG, LG, etc. |
アミノ酸配列の場合は、Prottest で決定されたモデルを使用。 のように実行して設定する。成功すると Setting Aamodelpr to Fixed(LG) Successfully set prior model parameters のようなメッセージが出る。 ( ) の中を WAG などに変えることで、違うモデルを指定可能。BLOSUM は blosum、JTT は jones を入れるようだ (3)。 |
Distribution model: |
これも Prottest の結果に従う。以下のようにして設定する。
|
広告
「あとがき」で当サイトを参考にしたと書いてくれているラノベです。Kindle Unlimited で読めました。ストーリーと文章が良く、面白かったです。
解析の実行
以下のようにすると、マルコフ連鎖モンテカルロ法による解析が始まる。この例では、10000 世代を指定している。
細かい指定方法はバージョンによって異なるが、文献 3 にも書かれているように、正確な結果を得るために重要なポイントは以下の 2 点である。
- 連鎖が収束してからサンプリングすること。
- 十分なサンプル数を確保すること。
連鎖の収束
バージョン 3.1 以降のMrBayes では、デフォルトでは 2 つの mcmc を同時に走らせ、複数の mcmc 結果が樹形空間内で似た位置に収束しているのかをテストしている (4)。
連鎖の収束は、Average standard deviation of split frequencies (ASDSF) で自動的に確認される。上記のコマンドで mcmc を開始すると、一定数ごとに以下のようなメッセージが出る。
Average standard deviation of split frequencies: 0.019298
マニュアルには、この値が
数字は基本的に単調に減少するが、増える場合もある。グラフは、300 万 generations 回してみたときの変化。増減を繰り返しながら、徐々に下がっていく感じ。
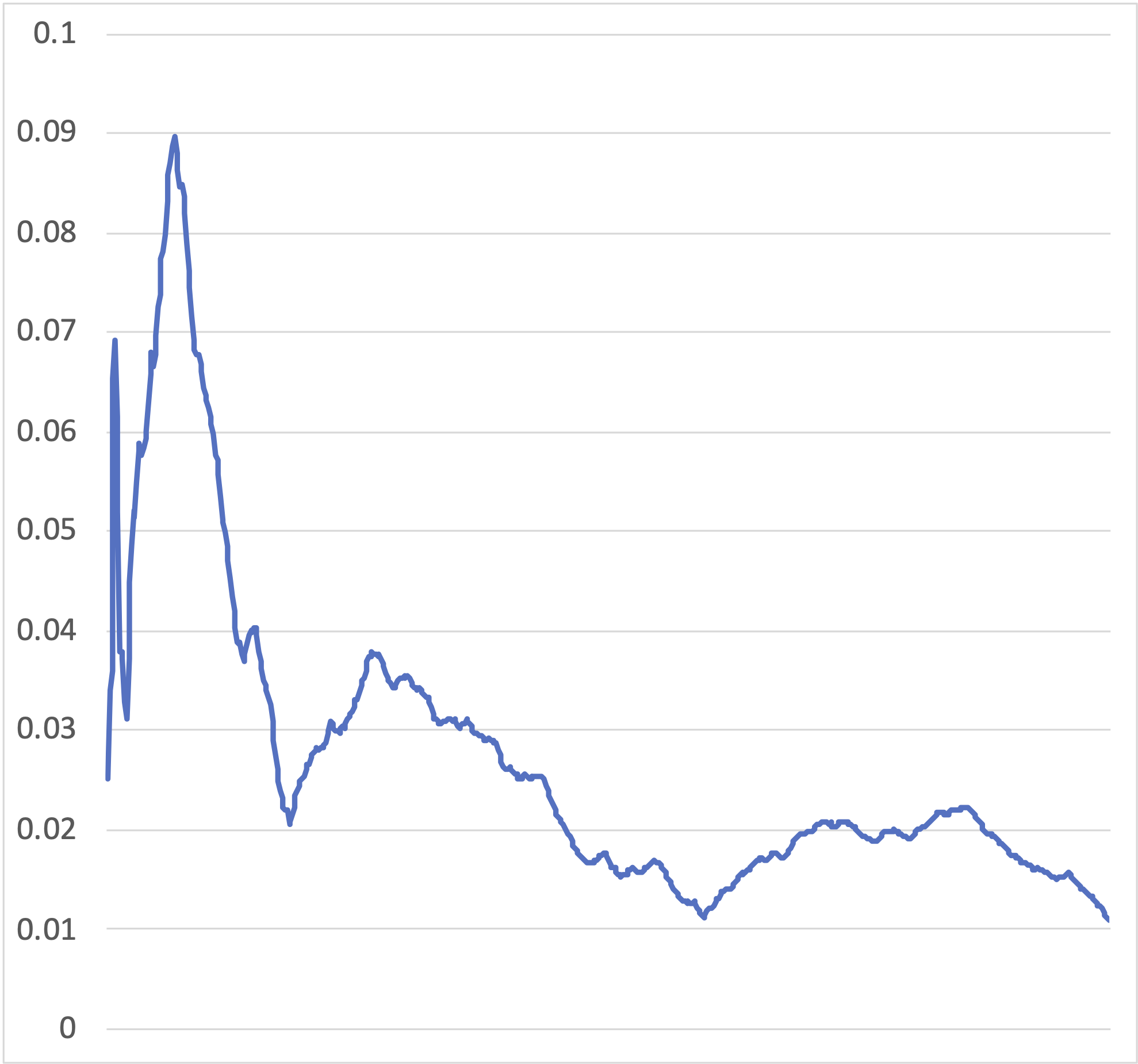
- データセットによっては収束しない場合もある。「局所最適解に捕まる」と表現されている (4)。
- 複数の mcmc を走らせることで、一つが局所最適解に捕まっても、他が最適解に収束していれば OK。
- したがって、mcmc は複数回走らせるか、8 以上にすることを勧めているページがある (4)。
解析の終了
0.01 以下になったら、
で結果を要約する。250 のところには、
mcmc ngen=10000 samplefreq=10 で解析を開始した場合、樹形のトータルが 10000, 10 につき 1 個を採用しているので、樹形数は 1000 である。その 1/4 なので、250 を入れることになる。Generation によって変更する必要があるので注意する。
この結果でチェックするのは、次の 2 点。
このようなグラフが出てくるので、ここに特定の増減傾向がないことを確認する。
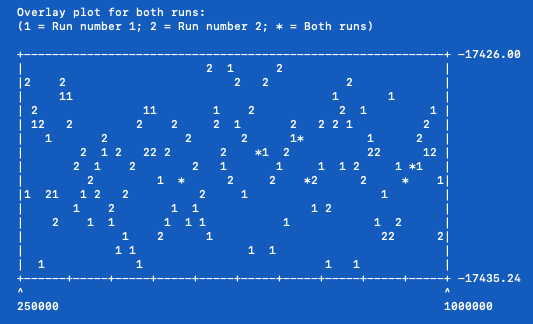
また、グラフの下に出てくる表に PSRF+ という値があり、これが 0.9 - 1.1 程度に収まっていること (3)。1.0 に近いほど良いらしい。
以上を確認できたら、
のようにして結果のファイルを出力する。数字の部分は sump と同じにする。つまり樹形数の 1/4 の数字を使う。
sample.nexus.con.tre のような名前のファイルが、系統樹のファイルである。sample.nexus の部分には、読み込んだ nexus ファイルの名前が入る。系統樹のファイルは、TreeView, FigTree のようなソフトや、IcyTree で可視化できる。
- sample.nxs.run1.p というファイルに、ターミナルウィンドウに表示される generation などのパラメーターが記録されている。何世代回したかをチェックしたいときは、このファイルを参照する。
解析を途中で止めたい場合
Average standard deviation of split frequencies: は一定世代ごとに表示されるので、始めにセットした世代数に到達する前に 0.01 を切ることもあるだろう。そのような場合は、
.t のファイルに end; をマニュアルで付け足すと解析できるようになるとしている ページ があるのだが、これをしても Could not open file "temp.run1.p" というエラーが出て sump ができなかった。確かに、結果のフォルダにそのファイルはないのだが、途中で停止せずに終了した解析にも temp で始まるファイルはない。
広告
References
- MbWiki Tutorial. Link: Last access 2022/01/08.
- Lset. Link: Last access 2022/01/08.
- MrBayes 3.1.2 を用いた系統解析. Link: Last access 2022/01/08.
- MrBayes Wiki. Link: Last access 2022/08/15.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。