次世代シークエンシング: 概要と用語集
広告
概要: 次世代シークエンシングとは
次世代シークエンシング (Next generation sequencing, NGS) とは、並列にシークエンスを行うことで高いスループットを達成した一連のシークエンシング技術である。
第二世代のシークエンサー
NGS は、ハイスループットを達成したシークエンシング技術の総称であるため、複数の原理がある。
非常に大雑把に言うと、2005 年頃から登場し、2013 年頃には比較的リードが長い Roche 454 系と、リードが短い Life Science の SOLiD、Illumina の HiSeq 系が競合していた。
このあたりの技術が、いわゆる「次世代」シークエンスという。サンガーシークエンスが第一世代で、その次の世代という意味。「第二世代」と呼ばれる場合もあるようだ (4)。
以下の表に概要をまとめた。どの技術も、
| Platform | リード長 |
その他特徴など |
|---|---|---|
Roche 454 GS FLX+ |
up to 1 kb |
Cited from this page. 原理は pyrosequencing。 |
Roche 454 GS Jr. |
up to 700 b |
Cited from this page. GS FLX の廉価版。 |
HiSeq 2000 |
50 b 前後 |
Ref 6, Illumina 社。 |
SOLiDv4 |
50 - 100 b |
Ref 6, Life Sciences 社 (旧 ABI)。 |
第三世代のシーケンサー
次世代シーケンサーは、非常に技術の移り変わりが早い分野である。
Roche 454 は最初に市場に出てきた次世代シークエンシング技術であったが、次第に使われなくなり、2016 年には Roche は市場から撤退した (4)。
かわって、2013 年ごろからメジャーになってきたのは、1 分子シークエンスを行う PacBio である。これはマイクロ PCR をベースとした上記の技術とは異なるので、第三世代とも呼ばれる。これにより、de novo アセンブリーなしにゲノム配列を決定することも可能になってきた。
| Platform | リード長 |
その他特徴など |
|---|---|---|
NovaSeq6000 |
アウトプットが TB のスケールに達した Illumina のモデル (4)。 |
|
PacBio RSII |
Single Molecule Real Time Sequencing (SMRT) により、数 kb のリード長を達成。 |
|
ナノポア |
膜タンパク質の微細構造を DNA 分子が通り抜ける際の電流変化で配列を読む。DNA ポリメラーゼに頼らない技術。第四世代? 解読長は 4 Mb を超えた (4)。 |
広告
「あとがき」で当サイトを参考にしたと書いてくれているラノベです。Kindle Unlimited で読めました。ストーリーと文章が良く、面白かったです。
NGS とバイオインフォマティクス
アウトプットは、50 bp から 1 kbp 程度までの塩基配列
リードの重ね合わせから得られる一続きの配列を
したがって、NGS の技術はコンピューターを使ったバイオインフォマティクスと高い親和性がある。
NGS 関連用語集
リード |
シークエンサーで決定した塩基配列のこと。NGS で読まれた配列に対して使われるのがほとんどである。 |
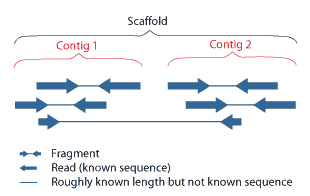
コンティグ |
複数のリードが連結されてできた一続きの配列。Contig には位置情報は含まれないが、scaffold には含まれる。 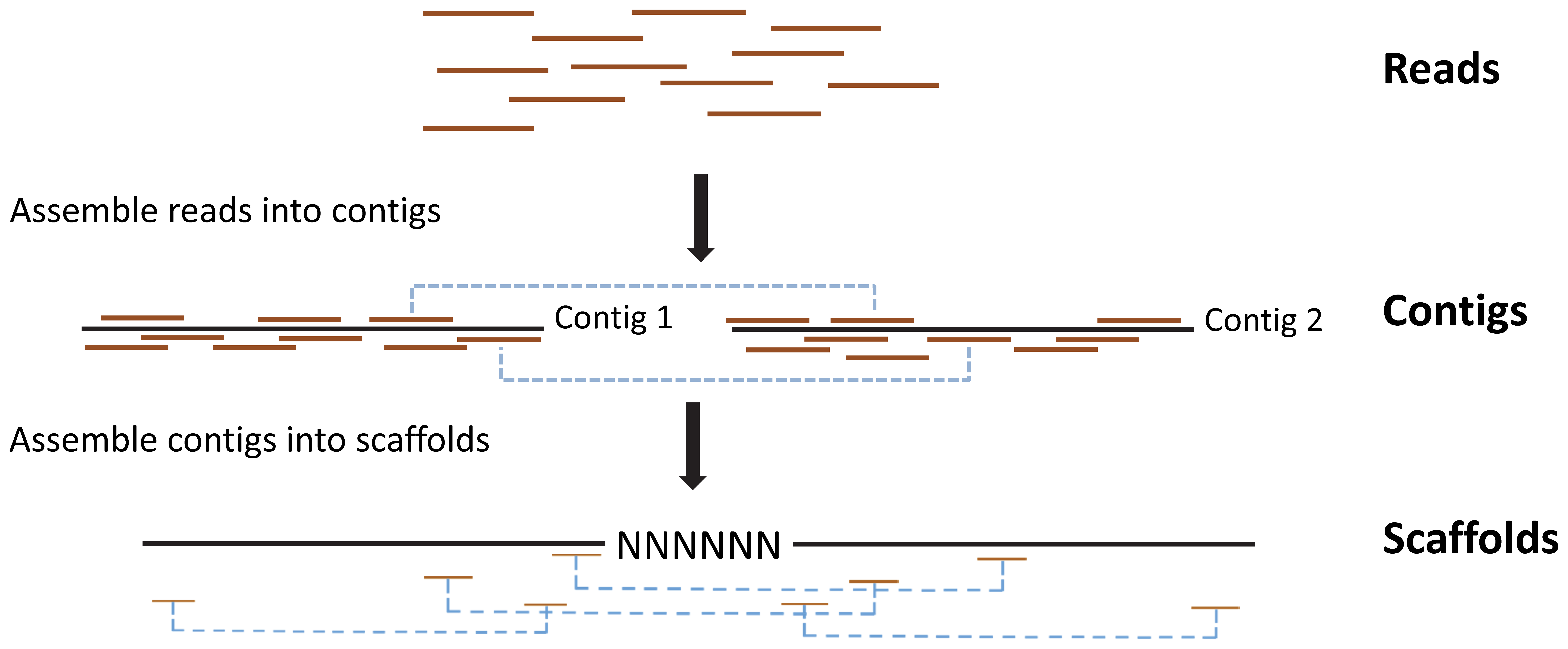
|
Scaffold |
コンティグ間の位置関係を示したもの。 |
N50, N60, etc. |
得られたコンティグを長い順に並べつつ、その長さを足していく。長さの合計が、総コンティグ長の 50% になったときのコンティグの長さを N50 という。アセンブリの質を示す値であり、高いほどよくアセンブルされたことになる。 総コンティグ長が長くても、短いコンティグが多ければ N50 も短くなる。N60, N80 などの値も使われることがある。 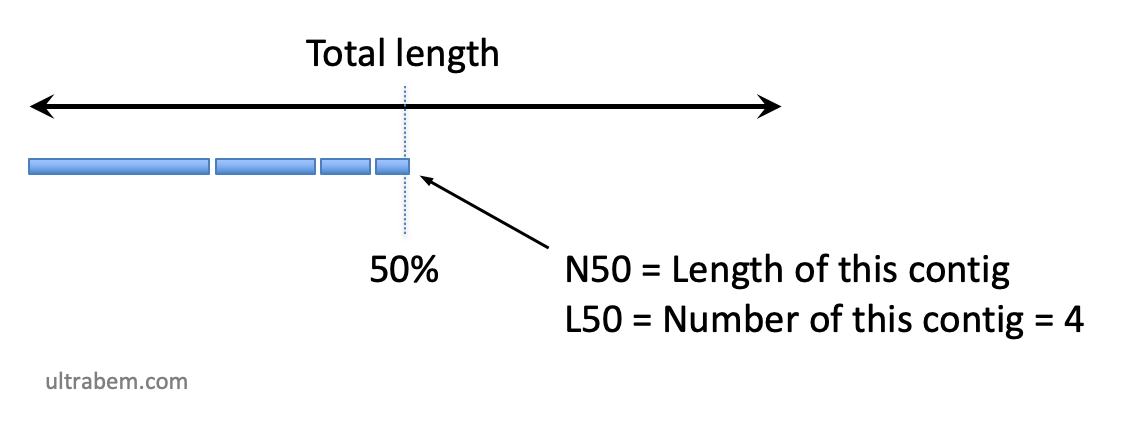
|
L50 etc. |
N50 と似ている。得られたコンティグを長い順に並べつつ、その長さを足していく。長さの合計が、総コンティグ長の 50% になったときのコンティグの |
Paired-end |
NGS で配列を決定するときに、一つの DNA 断片を片側からのみ読む場合と、両側から読む場合がある。前者を single-read sequencing、後者を paired-end sequencing という。また、これらの方法で得られたリードを single read, paired-end read という。 Paired-end read は、それぞれの配列自体がもつ情報に加えて、 |
k-mer |
ある配列があるとき、長さが k である部分配列を k-mer という。下の図 (5) では ATGG というもとの配列があり、TGG と ATG がその k-mer となる。TGG, ATG ともに長さは 3 なので、この図には 2 つの 3-mer が示されていると言える。 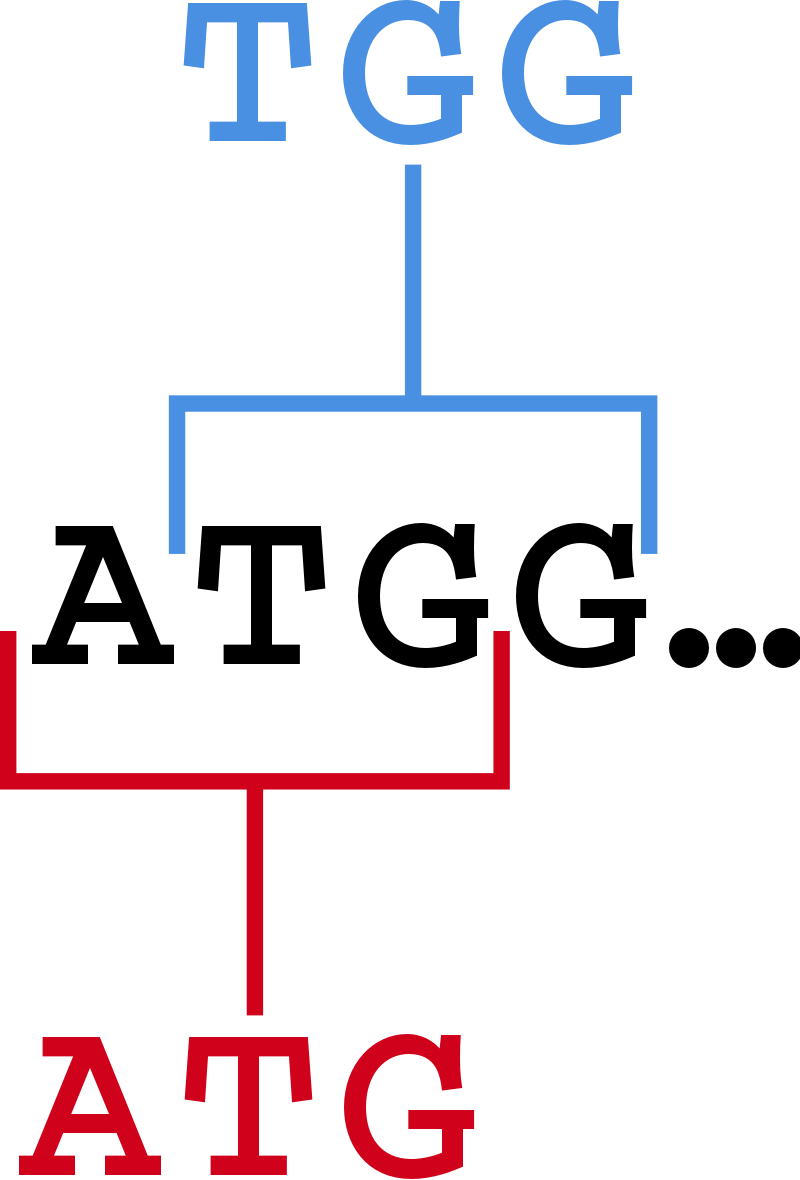
|
RNA-seq |
Transcriptome は転写産物の情報の総体、RNA-seq は実験手法。EST は NGS がメジャーになる前によく使われていた言葉で、transcriptome に相当するが、一般にデータ量は少なく、リードは長い。図は Griffith et al. (Ref. 4) 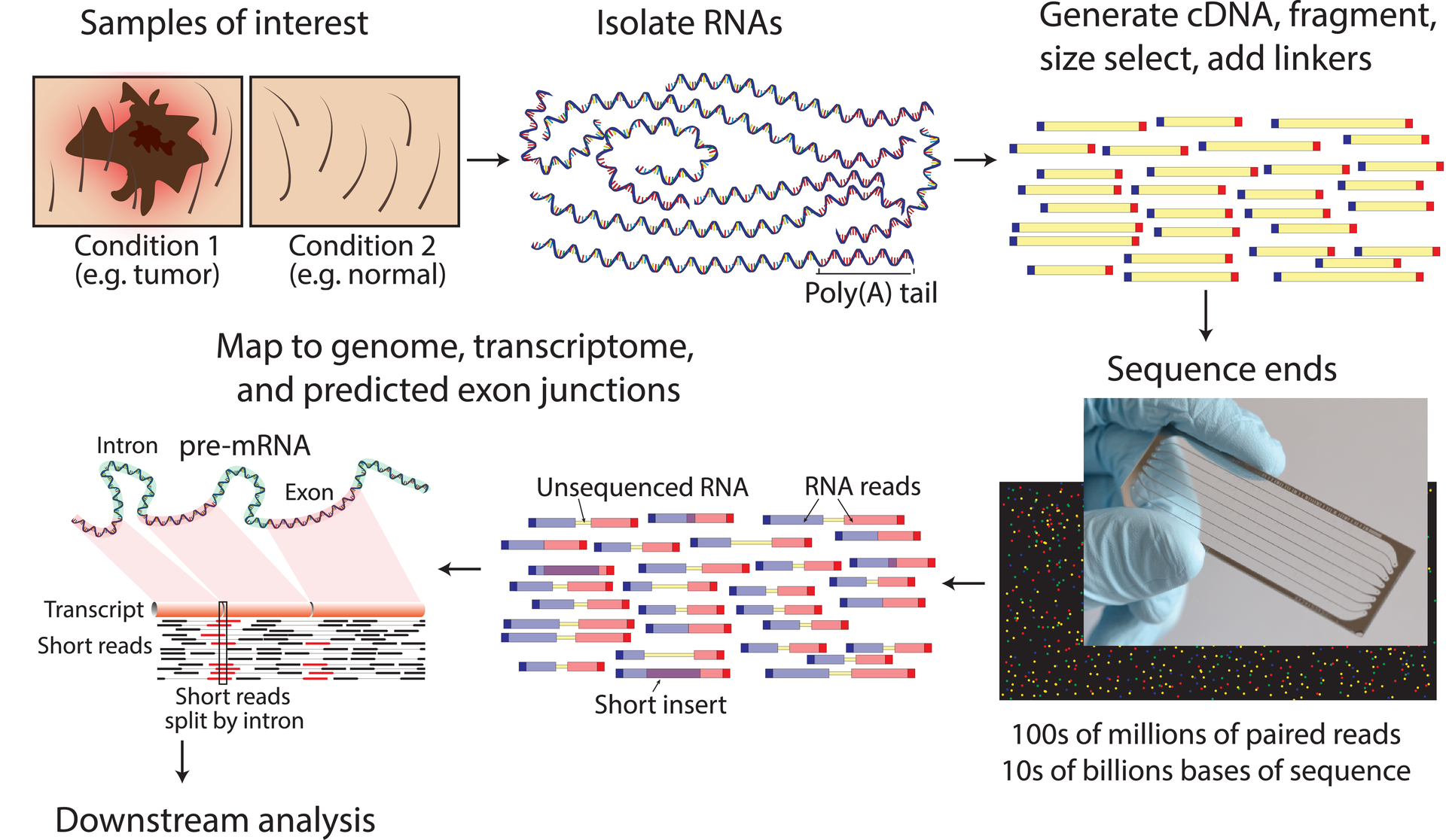
|
広告
References
ページ分割のため本文中に引用されていないものがありますが、番号と本文は対応しています。
- Amazon link:
Pevsner 2016. Bioinformatics and Functional Genomics.
- Amazon link:
清水、坊農 2019. 次世代シークエンサーDRY解析教本: 使っているのは第1版ですが、改訂第2版を紹介しています。
- Paired-End Read って何ですか? Link: Last access 2020/12/12.
中村 2021. 次世代シーケンス技術の現状と今後 - 2020. 生物工学会誌 99, 242-245.- By Ytngargar - Own work, CC BY-SA 4.0, Link
Liu et al. 2012a. Comparison of next-generation sequencing systems. J Biomed Biotechnol, 251364.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。