アセンブリープログラム cap3: インストールと実行
UB3/informatics/bioinformatics/cap3
このページの最終更新日: 2026/07/11広告
cap3 のインストール
cap3 は、重複部分のある配列をつなぎ合わせる
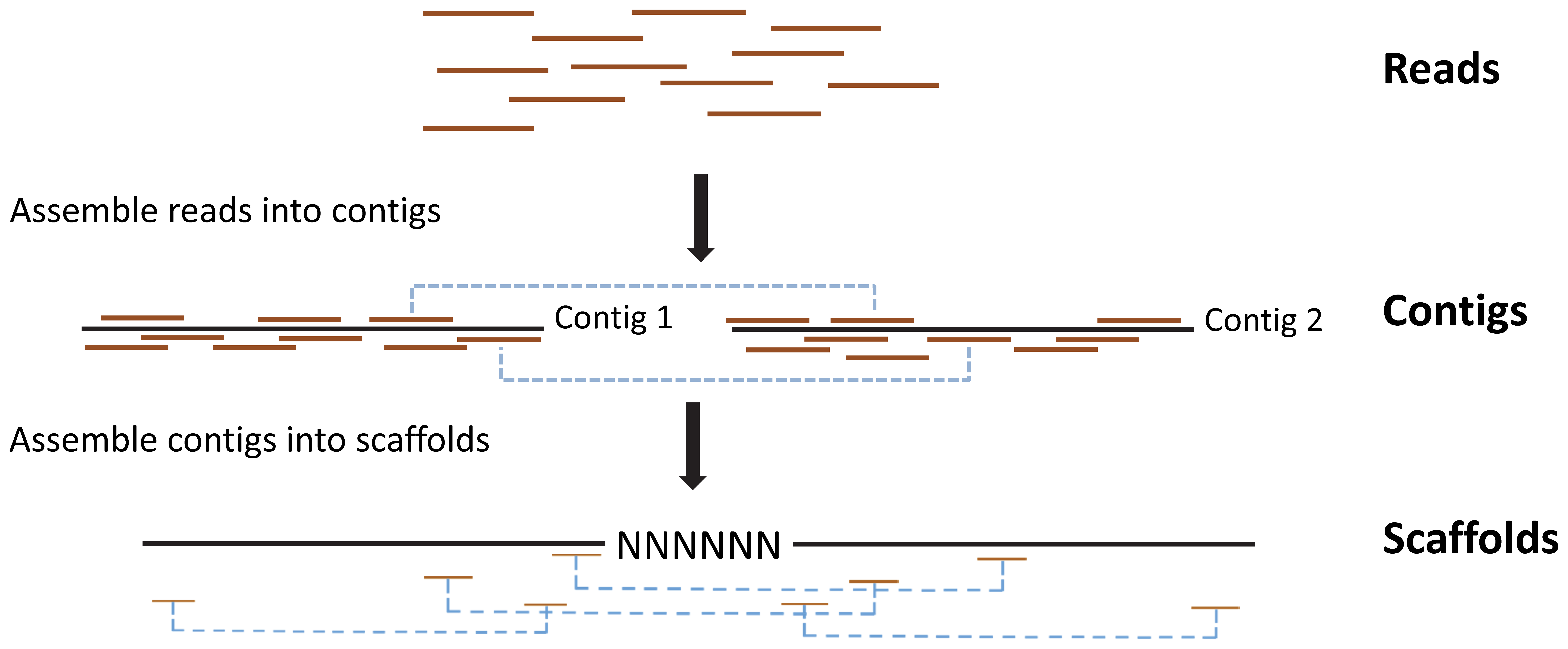
一般的事項および他の assembly program については、Assembly program の概要 のページを参照のこと。一応、ここにも assembly の概念図を載せておく (4)。
Mac へのインストール
cap3 は ここ からウェブベースで走らせることも可能である。しかしときどきサーバーが落ちていることがあり、またローカルで走らせた方が早いので、以下のようにしてインストールする。
2 通りの 方法を紹介しておく。
簡単なのは、homebrew を使って brew install cap3 とすること。環境によっては、brew install brewsci/bio/cap3 とする必要があるようだ。
もう一つは、ファイルをダウンロードして実行する方法。
- このページ から圧縮ファイルをダウンロードする。
- たとえば cap3.macosx.intel32 をダウンロード、解凍すると、同名のフォルダができる。
- この中に cap3 が入っているので、これを ターミナル で実行すれば良い。
ただし、ターミナルで単に cap3 とすると、基本的なプログラムが収められているフォルダ usr/bin/ で cap3 を探してしまい、プログラムが見つからないというエラーが出る。したがってパス指定が必要である。簡単な方法は、cap3 がインストールされているフォルダまで移動し ./cap3 としてプログラムを実行すること。./ がカレントフォルダを表す。
または、Mac でパスを通す のページを参考にしてパスを設定すれば、cap3 と打つだけでコマンドが使えるようになる。
Linux へのインストール
2022 年 6 月、conda install -c bioconda cap3 でインストール可能であった。Ubuntu 18.04。
広告
「あとがき」で当サイトを参考にしたと書いてくれているラノベです。Kindle Unlimited で読めました。ストーリーと文章が良く、面白かったです。
cap3 の使い方
使い方はシンプルで、input の fasta ファイルを指定するだけ。
オプションは以下。

アセンブルが成功すると、次のようなファイルが生成される。seq.fasta というファイルをアセンブルした場合、
- seq.fasta.cap.contigs - 生成したコンティグが収められている。
- seq.fasta.cap.contigs.links - ??
- seq.fasta.cap.contigs.qual - 数字の羅列、たぶんアセンブルの quality
- seq.fasta.cap.contigs.info - アセンブルのログみたいなもの
- seq.fasta.cap.contigs.singlets - アセンブルされずに残った配列
とりあえずは、contigs と singlets が配列の情報である。
input ファイルの大きさなど
cap3 は、基本的に小さい fasta ファイルのアセンブリーに使うプログラムである。メモリ使用量も、ABySS に比べると大きい。ネブラスカ大学のこのページ に有用な情報がある。自分の経験も足して、簡単に表にしておく。
Input file size |
Run time |
Used memory |
37 MB |
1.6 h |
1.5 GB |
150 MB |
2 h |
5 GB |
836 MB |
12 h |
28 GB |
1.5 GB |
Error |
64 GB 以上? |
1.5 GB の fasta ファイルを input にしたときには、エラーログ にある segmentation fault のエラーが出た (2022 年 6 月)。メモリ 64 GB の、通常よりは良いコンピューターだったのだが、かなりメモリを使うと思われる。
この 1.5 GB のファイルを seqkit split で分割してから cap3 するとちゃんと実行されるので、これはフォーマットの問題でなく、ファイルサイズの問題であると思われた。
配列を追加するとき
長い配列 (大きい遺伝子、mtDNA など) の全長を cap3 で決めようとするときの注意点。
たとえば最初のシークエンスで 7 個の配列があり、これをアセンブルすると Contig x 2, Singlet x 1 になったとする。
さらに実験をして得た 3 個の配列を追加してアセンブルしたいときは、
CAP3 エラーログ
segmentation fault (core dumped)
segfault として知られる一般的なエラー。あるソフトウェアが、アクセス禁止とされている領域 (アクセス禁止のメモリ領域、OS、read only 領域など) にアクセスしようとしたときに起こるようだ。
cap3 を使った場合のこのエラーが何を意味するかはわからないが、input data format に問題があったとする ページ がある。
広告
References
- de novo Transcriptome; 454用のベストなアセンブラーはどれだ!? Link: Last access 10/29/2017.
Kumar and Blaxter 2010a . Comparing de novo assemblers for 454 transcriptome data. BMC Genomics 11, 571.Huang and Madan 1999a . CAP3: A DNA sequence assembly program. Genome Res, 9, 868-877.Johnson et al., 2012a. Evaluating methods for isolating total RNA and predicting the success of sequencing phylogenetically diverse plant transcriptomes. PLoS ONE 7, e50226.
Figures are cited from open-access articles distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。