メモリ使用量の低いアセンブリープログラム ABySS:
インストール、使い方など
UB3/informatics/bioinformatics/abyss
このページの最終更新日: 2026/04/09広告
概要: ABySS とは
AbySS は short paired-end reads のための de novo assembler である。アセンブリー全般については、アセンブリーの概要 を参照のこと (図、文献 2)。ABySS については GitHub のページ(6) が本家である。
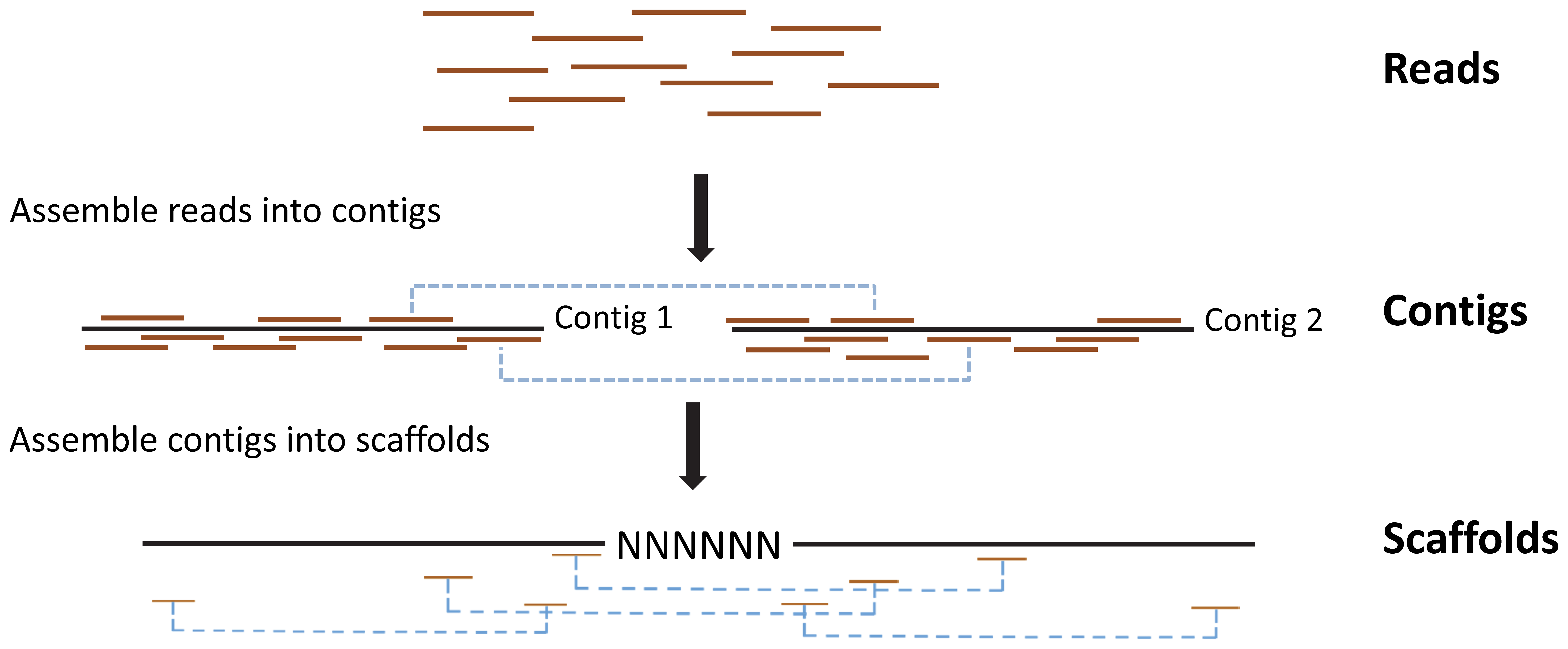
ABySS 1.0 は、SOAPdenovo などと同様に 600 GB ものメモリを必要としたようだが、ABySS 2 では bloom filter という方法を使うことで
35 GB でヒトゲノムのアセンブリができたと論文には記載されている (1,4)。オリジナル論文は文献 4。
ABySS のインストール
AbySS は Mac ならば brew または conda、Linux ならば apt-get install または conda でインストールできる。conda install -c bioconda abyss。
ABySS の使い方
Paired-end のアセンブリーは以下のようにする。k は k-mer の長さ、ペアリード名には 1 と 2 の数字が入っていないといけない。name はアウトプット名で、生成されたコンティグは name-contigs.fa などという fasta ファイルに保存されるようだ。
k-mer の設定
k=96 のようにして、k-mer を指定する。大まかな目安は以下の通り。
- 2 x 150 bp, 40x coverage の場合、適切な k-mer は 70 から 90 である (6)。50 から 90 の間で、for loop で試すスクリプトが載っている。
- デフォルトの k-mer 最大値は 192 (6)。
- k についての参考になるかもしれない ページ。
使用メモリの設定
ABySS は、Bloom filter という方法で使用メモリを低く抑えることに成功したアセンブラーである。この Bloom filter mode は、B=2G のようにして使用するメモリを指定することで on にできる (6)。
Bloom filter を使わない MPI モードに比べると、使用するメモリは約 1/10 になるようである。文献 7 では、約 3 Gbp のヒトゲノムのアセンブリーに必要なメモリは、Bloom filter を使わないと約 1 TB、使えば約 30 Bb と書かれている。
「指定したメモリ量の 9 分の 8 が自動的に counting Bloom filter に、9 分の 1 がもう一つの k-mer を track する Bloom flter に割り当てられる」とマニュアルに書かれている (6) が、ここはよくわからない。
最適なメモリ量は様々な要因に左右されるが、大まかな目安は以下の通り。
- ゲノムサイズ 約 20 GB, B=500G
- ゲノムサイズ 約 3 GB (ヒトゲノムのサイズ), B=50G
- ゲノムサイズ 約 100 M (C. elegans のゲノムサイズ), B=2G
単位には k, M, G が使用できる。普通は G を使うだろう。大きな数を指定しても、実際にその量のメモリが使われてしまうこと以外のデメリットはない。
メモリ使用量などをモニターできる top コマンドで、B=50G のときの状態を見てみたスクリーンショットが以下。
全体では 64 GB, そのうち 45 G ほどが使われており、その 70.9%、つまり 31 GB 前後のメモリが ABySS に使われていることがわかる。
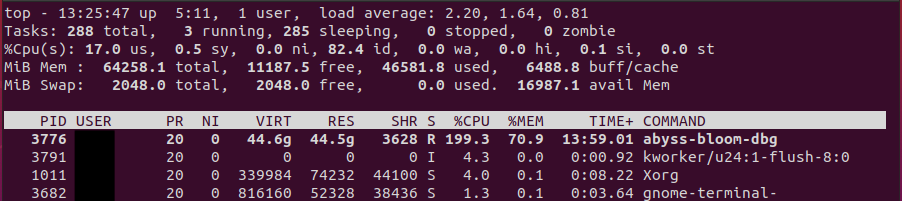
これを B=60G にすると、全体の使用量と ABySS のパーセンテージがともに上昇し、free が 400 Mb ぐらいしかなくなる。クラッシュを避けるためには、あまり多くを割り当てすぎない方がいいのか?
文献 7には、使用メモリを減らす方法として、以下のような点が挙げられている。
- k を増やす
- kc (minimum k-mer occurrence cutoff) を増やす
- m=k-1 をコマンドに加える。これはよくわからないので、コメントをそのまま書いておくL ... to identify `AdjList` to only find k-1 overlaps. Normally `AdjList` finds overlaps between 50 bp and k-1 bp, but finding overlaps less than k-1 bp requires building a suffix array which in turn requires more memory (`AdjList` builds a compacted de Bruijn graph by finding end-to-end overlaps between pairs of sequences.)
ABySS の output
- Unitigs: Paired-end 情報を使わずにアセンブルされた配列。Single-end data を使った場合、これが唯一の結果ファイルとなる。contigs や scaffold も出る場合があるが、ファイルの中身は unitigs と同じだった。
- Contigs: Paired-end 情報を使ってアセンブルされた配列。Contains sequences assembled with paired information, scaffolding over sequencing coverage gaps, but no repeats.
- Scaffolds: Contains sequences assembled with paired information, scaffolding over sequencing coverage gaps and repeats.
- Long Scaffolds: Contains sequences that were obtained by rescaffolding using long sequences libraries.
- **-stats.csv というファイルに、abyss-fac で得られるコンティグ情報が一覧になっている。
2022 年 5 月、unitigs はシンボリックリンクで与えられ、output-3.fa というファイルにリンクが張られていた。-1.fa, -2.fa というファイルもあった。input 配列によっては、-8.fa ぐらいまで行ったことがある。
このページ によると、ABySS は複数のステップでアセンブリーし、そのたびに番号を増やしていくので、この場合は output-3.fa のみが最終結果であり、他のものは通常は必要なさそう。また、.dot などの拡張子をもつファイルも作られるが、これも通常は必要ないようだ。
2022 年 4 月に GitHub ページの例を試してみた結果。まず以下のようにテストデータセットをダウンロードして解凍する。
tar xzvf test-data.tar.gz
これをアセンブルすると、test-contigs.fa や test-scaffolds.fa などのファイルが作られる。
Contiguity statistics を計算する。n, n:500, L50, min, N75, N50, N25, E-size, max, sum, name のパラメーターが表示される。
assembly-stats というプログラムを利用してもよさそうだが、これは新たにインストールする必要があるので、abyss-fac を使う方が便利だろう。
ただし、abyss-fac は 500 nt 以下の contig を結果に含めない設定になっている (参考) ので注意。同じ fasta ファイルに対して seqkit stat をすると短い配列も含まれるので、これは abyss のアウトプットでカットされているのではなく、statistics を表示するときに省かれている。
広告
「あとがき」で当サイトを参考にしたと書いてくれているラノベです。Kindle Unlimited で読めました。ストーリーと文章が良く、面白かったです。
広告
References
- Mac でインフォマティクス ABySS 2.0. Link: Last access 2021/04/23.
Johnson et al., 2012a. Evaluating methods for isolating total RNA and predicting the success of sequencing phylogenetically diverse plant transcriptomes. PLoS ONE 7, e50226.- ABySS, Genome Science Center.Link: Last access 2022/04/23.
Jackman et al., 2017a. ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter. Genome Res27, 768-777.- ABySS. Omicsbox. Link: Last access 2022/04/23.
- GitHub のページ: Last access 2022/05/09.
- Google conversations. Link: Last access 2022/09/04.
Figures are cited from open-access articles distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。