R による LogFC (fold change) の計算
UB3/informatics/r/logfc
このページの最終更新日: 2026/07/11広告
概要: LogFC とは
RNAseq、プロテオミクス、メタボロミクスなどの大規模解析では、しばしば
この fold change を対数で表した
「常用対数 log と自然対数 ln の違い」に、対数に関する基本事項をまとめてあります。以下のサイト内検索からどうぞ。
広告
「あとがき」で当サイトを参考にしたと書いてくれているラノベです。Kindle Unlimited で読めました。ストーリーと文章が良く、面白かったです。
2 群で LogFC を計算する
組み込みデータセット swiss を使った例を載せておく。
swiss は、以下の図 (一部のみ表示) のようなデータセットである。スイスの各地域の出生率、Education などがまとめられている。
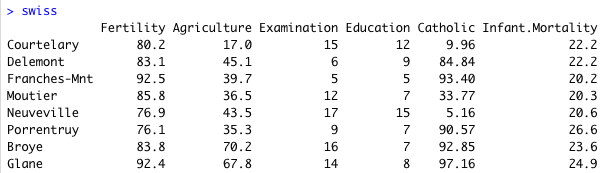
ここでは、出生率 fertility を、80 以上なら 1、80 未満なら 0 とし、0 または 1 で Agriculture, Examination, Education, Catholic, Infant.Mortality の LogFC を比較する。
具体的な手順は以下の通り。
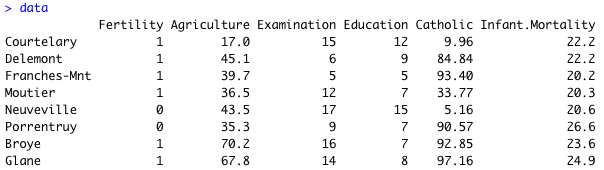
1. Fertility を 0/1 データに変換
data$Fertility[data$Fertility < 80] = 0
data$Fertility[data$Fertility >= 80] = 1
これで Fertility が 0 と 1 に変換される。
2. 変換した 0 と 1 でグルーピングする
Fertility が 0 の群と 1 の群に分ける。Excel などで並べ替えても良いのだが、dplyr に group_by という便利な関数があるので、これを使ってみる。
これで、data を Fertility の 0/1 によって 2 つの群にわけることができる。この関数の詳細は、このページ などを参照。
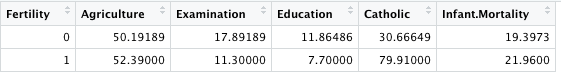
このあとに data の内容を表示しても表示してもソートされているわけではないが、グループ分けはされている。確認するためには、summarise_all 関数などで計算してみるとよい。
B = summarise_all(A, funs(mean(., na.rm=TRUE)))
全てのグループについて、mean 関数で平均値を計算している。na.rm = TRUE は欠損値を無視するオプション。すると、このように 0 と 1 ごとに平均値が計算される。
3. 整然データに変換し、LogFC を計算
これらの値の比をとれば fold change であり、その対数をとれば良いのだが、この形は
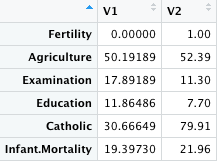
ただし、t 関数で変換するとデータフレームではなくなってしまうため、as.data.frame 関数で D に戻す。D はこのようなデータフレームになる。
D = as.data.frame(C)
新たな列 V3 を cbind 関数で追加する。as.data.frame を使わないと上手くいかないのは、ここも同じである。
F = cbind(D, E)
最後に V3 を Log にする。これで一応完成。
H = cbind(F, G)
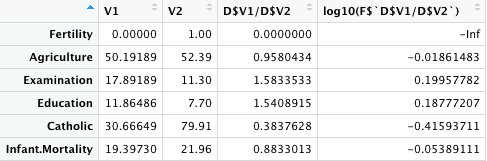
しかし、この方法では Fertility の行が -Inf になってしまう。ゼロの対数をとっているためである。そもそも、この行は 0 または 1 の条件の行なので、平均値などを計算することに意味はない。データフレームから特定の行を抽出する に従って、2 - 6 行目だけ抽出すれば十分なはず。
以上で、これで複数の値に対して一括で LogFC を計算できたことになる。今回は各ステップを見るためにいちいち変数に格納したが、慣れてきたら一気にパイプ %>% で繋いでしまって大丈夫だろう。
LogFC の計算でゼロを避ける方法
Fold change および LogFC の計算方法は、ゼロがない場合にのみ通用する方法である。
最後の段階で、V1 の値にゼロがあれば、Fertility の行のように Log100 を 計算しなければならなくなる。10 を n 乗してゼロになる数は計算できないので、LogFC ではこれを避ける必要がある。
V2 の値にゼロがあれば、V1/V2 の部分でゼロで割ることになってしまう。Fold change でも LogFC でも問題となる。
これを避けるために、以下のような方法がとられている。
- RNAseq のリード数をデータとして使うときは、最初に閾値を設定する。RPKM > 5 などの基準を設けて、それよりリード数が少ない遺伝子は除外 (1)。
広告
References
- fold change for genes with 0 reads. Link: Last access 2022/04/28.
- 遺伝子発現量などのカウントデータで倍率変化 (fold change)を求めるとき. Link: Last access 2022/04/28.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。