Decipher によるシンテニー解析
UBC/informatics/r/select
このページの最終更新日: 2026/07/11広告
Decipher の使い方
Decipher の instruction は 文献 1 に。他のプラットフォームでも動くのかもしれないが、ここで説明するのは R で実行する方法。まず、Biocmanager でインストールする。
解析の実行
サンプルコードも、文献 1 の Alignment – Align Synteny のところに載っている。少しわかりにくいところもあったので、自分が使ったものを下に載せておく。
ライブラリの読み込み後、比較したいファイルへのパスを fas という変数に格納する。例では 3 個の配列が使われているが、現実的な個数なら何個でも大丈夫。
fas <- c(Genome1 = "./genome/genome1.fasta",
Genome2 = "./genome/genome2.fasta",
Genome3 = "./genome/genome3.fasta")
次に、データベースへのパスを指定する。decipher では、MySQL のようなデータベースに配列を入れてから解析するようだ。
もとのチュートリアルには、単に「db にデータベースへのパスを書く」と書いてあるのだが、これは以下のようにする。
このページ を参考にした。よくわかっていないのだが、もしかするとこれはメモリの中に作業用のデータベースを作るということで、そのために後述するようにメモリ使用量が大きめなのかもしれない。
続いて、for loop を使って配列をデータベースに読み込む。これは元のコードから変更なし。
Seqs2DB(fas[i], "FASTA", db, names(fas[i]))
}
FindSynteny 関数で、シンテニー解析を行う。数百 Mb から 1 Gb 程度の配列 2 個で、13 Gb 程度までのメモリを使い 10 分ほどかかる印象。低スペックなコンピューターだとクラッシュするかもしれない。
FindSynteny 関数で、シンテニー解析を行う。数百 Mb から 1 Gb 程度の配列 2 個で、13 Gb 程度までのメモリを使い 10 分ほどかかる印象。低スペックなコンピューターだとクラッシュするかもしれない。
結果の表示
pairs(synteny) #ドットブロットの表示
plot(synteny) #棒グラフの表示
Ensembl からいくつかゲノムデータを拾って比較してみた。ここでは、C. elegans と D. melanogaster を比較した結果を載せておく。
2022 年 9 月に実行。使用データセットは Caenorhabditis_elegans.WBcel235.dna.toplevel.fa.gz と Drosophila_melanogaster.BDGP6.32.dna.toplevel.fa である。
まずはテキストのサマリー。C. elegans と D. melanogaster は、0.43% しか align されないということか?
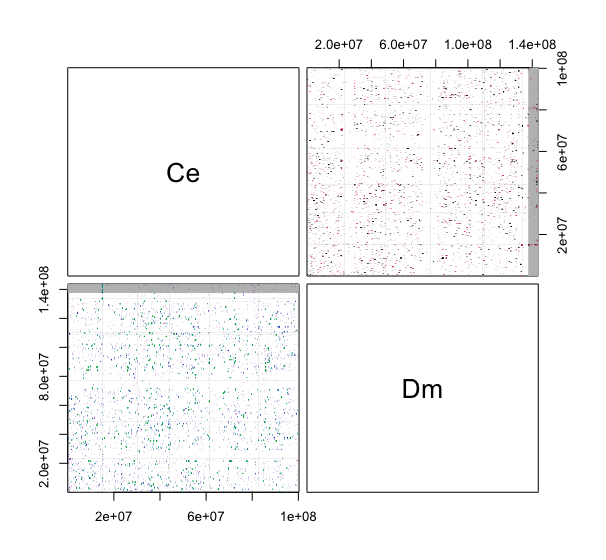
プロットするとこのようになる。解析データ: Emsembl にあるゲノムのシンテニー解析 という項目に他の比較データをまとめたので、そちらを参照するとわかりやすいが、C. elegans と D. melanogaster の比較では、多数の小さなドットのみが見え、ゲノムレベルでシンテニーはほとんど保存されてないと言えるだろう。
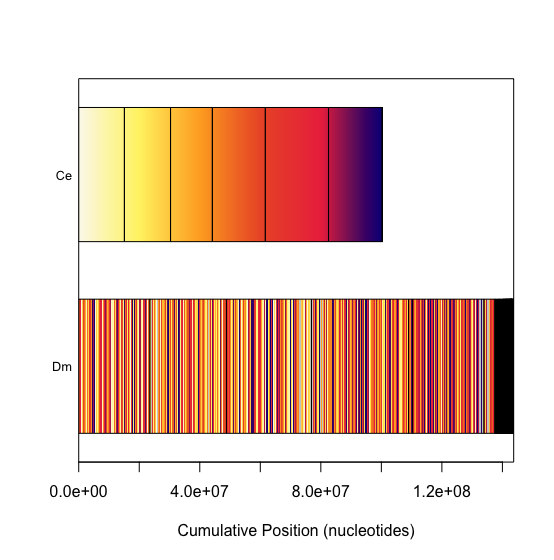
bar plot にしたもの。色がマッチしている部分が対応部分。D. melanogaster で、似た色が続く領域が出てくれば、そこが保存領域なのだが、あまり見当たらない。
広告
「あとがき」で当サイトを参考にしたと書いてくれているラノベです。Kindle Unlimited で読めました。ストーリーと文章が良く、面白かったです。
解析データ: Emsembl にあるゲノムのシンテニー解析
いろいろ遊んでみてから更新の予定。
広告
References
- Link: Last access 2022/09/04.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。