KEGG: 主に論文の図の作り方
UB3/informatics/bioinformatics/kegg
このページの最終更新日: 2025/11/23広告
概要: KEGG とは
KEGG は Kyoto Encyclopedia of Genes and Genomes の略であり、遺伝子やタンパク質の関係をデータベース化したものである。1955 年に、京都大学の金久實らによって始められ、世界中で広く用いられている。
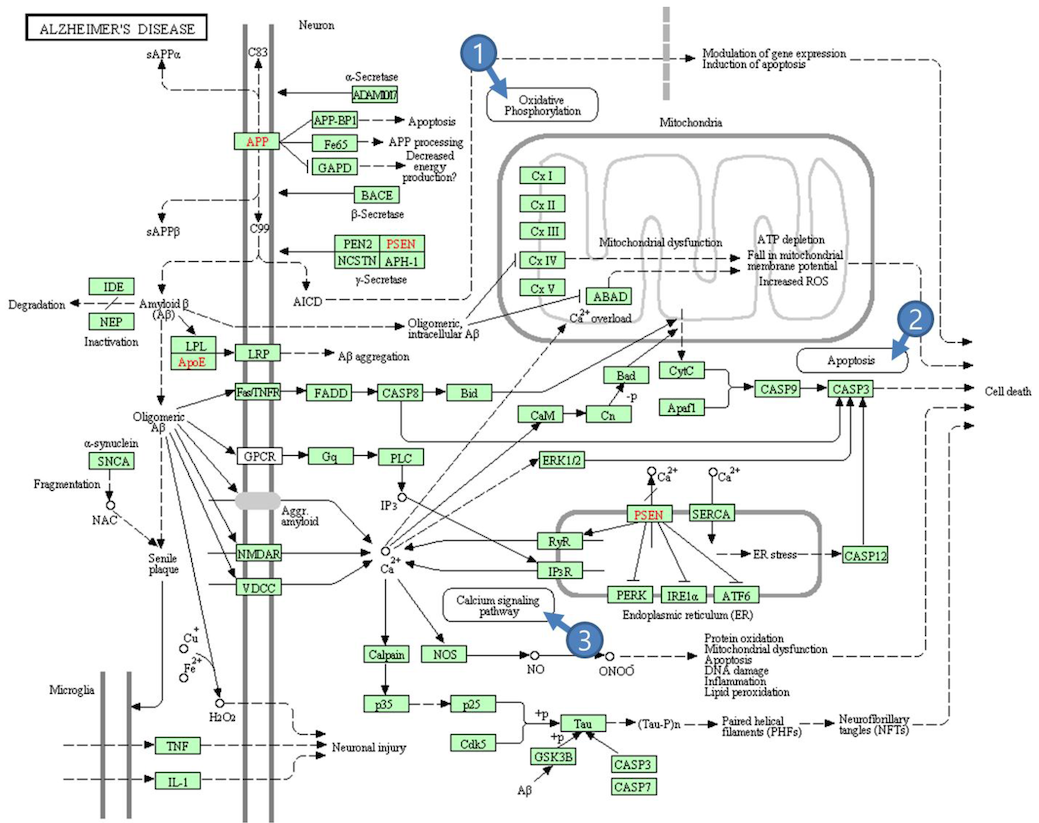
代表的な KEGG pathway は、例えば以下の図のようなものである。Alzheimer's disease というパスウェイ (5)。
KEGG orthology (KO) とは
KEGG orthology (KEGG オルソロジー) とは、ortholog に関連する KEGG のデータベースの一つで、KEGG pathtay の各ノードに対応した ortholog を定義したものである。もう少しわかりやすく言うと、遺伝子の一つ一つに KO1234 のような番号が付けられていて、遺伝子と KEGG pathway を対応させやすくしたもの。
例えば KO1601 はピルビン酸デヒドロゲナーゼの遺伝子であり、これは map00010 の Glycolysis / Gluconeogenesism、map00020 の Citrate cycle、map00620 の Pyruvate metabolism などに属している。
KO と map は 1:1 対応ではないことに注意。一つの遺伝子は、基本的に複数の map に属している。
Seq2Fun version2 パッケージのページに、KO に関する以下のような記述があった。このパッケージは fastq file を input とするようだ。
|
タンパク質コード遺伝子の一部しか KO が割り振られていない。例えばヒトでは ~ 19,648 の protein-coding genes 中、14,964 (76.16%) のみ。ゼブラフィッシュでは ~ 26,584 遺伝子中 16,322 (61.40%) のみ。 Pathways まで割り振られている遺伝子はさらに少ない。Gene annotation も限られている。 |
KEGG enrichment とは
KEGG のデータを扱う上で難しいのが「エンリッチメント」enrichment という概念およびエンリッチメント解析である。
エンリッチメント解析は、ある遺伝子リストに対して
- 2 つのヒト集団 A, B で遺伝子発現パターンを比較し、有意に発現量が異なる遺伝子が 1000 個得られたとする。
- 1000 個の遺伝子のうち、100 個が糖質代謝に関わる遺伝子であった (そのような KEGG pathway に割り当てられていた)。全体の 10% である。この結果には意味があるか?
- これは、ヒトの遺伝子中にどれだけ糖質代謝関連の遺伝子があるかという話になる。
- ヒトの遺伝子を仮に 25000 個として、2500 個 (10%) が糖質代謝に関わっていると仮定する。この場合、ランダムに遺伝子を 1000 個選んでも、10% は「糖質代謝に関わる遺伝子」ということになる。
- よって、A と B で差がある遺伝子 1000 個中、「10% が糖質代謝に関わる遺伝子」であるということに、ほとんど意味はないと考えられる。
- もし、「20% が糖質代謝に関わる遺伝子」であった場合、全遺伝子での割合に比べて
2 倍濃縮されている ことになる。この結果から、「A と B は糖質代謝に違いがある」と推察するのは合理的であろう。 - エンリッチメント解析とは、統計的にこの「濃縮度」を評価する解析である。
R でエンリッチメント解析を行う方法を、R による KEGG エンリッチメント解析 のページにまとめた。
広告
「あとがき」で当サイトを参考にしたと書いてくれているラノベです。Kindle Unlimited で読めました。ストーリーと文章が良く、面白かったです。
広告
References
Yu et al., 2012a. Yu G, Wang L, Han Y, He Q (2012). clusterProfiler: an R package for comparing biological themes among gene clusters.” OMICS: A Journal of Integrative Biology, 16, 284-287.Wu et al. 2021a. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141.- ページ編集に伴い削除
- ページ編集に伴い削除
Seo et al., 2015a. Development of network analysis and visualization system for KEGG pathways. Symmetry 7, 1275-1288.- エンリッチメント解析の基本. Link: Last access 2022/06/16.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。