塩基配列アラインメントの原理と主なプログラムの特徴
UB3/informatics/bioinformatics/alignment_programs
このページの最終更新日: 2023/02/14- Pairwise alignment の定義と原理
- スライド
- ギャップの挿入
- シャッフル
- 主なアラインメントプログラムの特徴
- どのアラインメントプログラムを使えばいいのか?
- 主なアラインメントプログラムの一覧表
- コマンドラインを使って Clustal Omega を走らせる
広告
Pairwise alignment の定義と原理理
塩基配列、アミノ酸配列などを特定の規則に従って並べることをアラインメント alignment という。アラインメントされた配列は、図 (Public domain) のような形で表されることが多い。

2 つの配列のアラインメントは (Amazon link, ref. 4) では、ペアワイズアラインメントは以下のように定義されている。
|
The process of lining up two sequences to achieve maximal levels of identity (and conservation, in the case of amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology. |
つまり、アラインメントの際には、maximal levels of identity を達成することが必要である。これは、同じアミノ酸を可能な限りマッチさせるということである。原則として以下の作業が行われ、最適なアラインメントが決定される。
スライド
2 つのアミノ酸配列を順にスライドさせていき、同一なアミノ酸の数を数える。
GDCEFHIJ
この状態では 3 番目の C だけが同一であるが、次のようにスライドさせることで D, E, F と 3 個の同一な文字が現れる。こちらの方がより良いアラインメントと言える。
GDCEFHIJ
もちろん、計算はアミノ酸が単に同一か異なるかだけではなく、アミノ酸の構造や機能の違いによって置換にスコアがあり、そのスコアを最大にするようなアラインメントが良いアラインメントとなる。
ギャップの挿入
下のように 1 個
GDCEFHIJ
ただし、無制限にギャップを導入できるとアラインメントが成り立たなくなるため、通常
シャッフル
得られた結果が偶然ではないことを確認する作業である。
配列の一方をランダムにシャッフルして、スコアの計算を繰り返す。すると、スコアは山形の確率分布を示すことになるだろう。すると、
この考え方は一般的な統計検定の考え方と共通し、ブートストラップにも少し似ている。以下のページも参照のこと。
主なアラインメントプログラムの特徴
どのプログラムを使えばいいのか?
もちろん、「これを使えば全て OK」という単純な答えはなく、
- 配列による
- 複数のアラインメントプログラムを試し、結果が一致するなら信頼性が高い
というのが回答である。しかし、それぞれのプログラムを比較した論文もたくさん出ているので、その結果をメモしていくことにする。
> Benchmark データセットを使い、9 つのプログラムを比較。2014 年 (7)。
- CLUSTALW, CLUSTAL OMEGA, DIALIGN-TX, MAFFT, MUSCLE, POA, Probalign, Probcons and T-Coffee を使用。
- Probcons, T-Coffee, Probalign, MAFFT が全体に正確であった。
- N, C 末端に大きな伸長配列がある場合は、Probalign, MAFFT, Clustal Omega が Probcon および T-Coffee よりも正確であった。
主なプログラムの一覧表
| 名前 | 特徴 |
|---|---|
| Clustal Omega | かつては Clustal W というプログラムがよく使われてきたが、2015 年 10 月現在では、EMBL のサーバーは Clulstal Omega を提供するようになっている (2)。 Clustal Omega の元論文は、おそらく Sievers et al. (2011)。オープンアクセス論文で、2018 年 10 月現在で 9000 回以上引用されている。 |
| Clustal W | Command line tool は 2021 年でもインストール可能。よく覚えていないが、brew で出たメッセージに従ったはず。 |
| MUSCLE | MUSCLE (MUltiple Sequence Comparison by log-Expectaition) は、2004 年に発表されたアラインメントプログラムである (1)。Clustal W よりも早く、オプションを適切に選べば原則として Clustal W よりも「良い」アラインメントを返すと主張されている (1)。 Command line tool もある (Website)。brew で簡単にインストールできた。 |
| T-Coffee | コーヒー系のバリエーションが多数あり、例えば Expresso ではタンパク質構造の情報もアラインメントに取り入れているようだ。オリジナルは文献 6 の Notredame et al., 2000 である。 |
| M-Coffee | You can use T-Coffee to align sequences or to combine the output of your favorite alignment methods (Clustal, Mafft, Probcons, Muscle, etc.) into one unique alignment (M-coffee) という説明があり、含めるプログラムをオプションで選択できる。 Muscle や Probcons も含まれているようである。 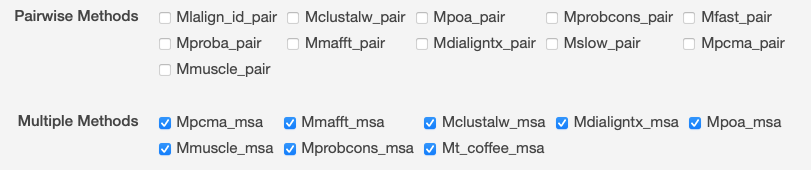
文献 は T-Coffee と同様に Notredame et al., 2000。 |
| MAFFT | 2022 年 3 月現在、このリンクで version 7 が使える。 |
| Probalign | 2022 年 3 月現在、このページが生きているが、最終更新が 2010 でメンテナンスされている形跡がないのが気になる。 |
| ProbCons | これもあまりメンテナンスされている形跡なし。 |
コマンドラインを使って Clustal Omega を走らせる
以下の作業が、2018 年 3 月 1 日時点、Mac High Sierra で有効であった。
- このページ からダウンロード、名前を clustalo に変えて好きなフォルダに置く。
- chmod +x clustalo を実行し、プログラムを実行する権限を追加。
- そのフォルダで、FASTA 方式で保存した テキストファイル を読み込んで実行。
./ clustalo -i seq.txt --
./clustalo --help でヘルプを参照することができる。-i で読み込むファイルを指定。
Output format は表現が少しわかりにくい。
--outfmt={a2m=fa[sta],clu[stal],msf,phy[lip],selex,st[ockholm],vie[nna]} MSA output file format (default: fasta)
とヘルプに書かれている。これは、clustal format でアウトプットしたいときは
./clustalo -i test.txt --outfmt=clu
のように書けということ。
|
|
|
|
広告
References
Edgar 2004a. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–97.- Clustal Omega. Link.
- Amazon link: Berg et al. Biochemistry: 使っているのは 6 版ですが 7 版を紹介しています。
Pevsner, 2016a. . Bioinformatics and Functional Genomics, 3rd ed.
|
2016 年の出版で、この分野ではもはや古い部類に入る教科書かもしれないが、データベース検索、アラインメントの基礎が多くのわかりやすい図とともに述べられている良書。 Bioinformatics に欠かせない Linux コマンドも、基礎から丁寧に解説されている。結果としてかなり厚い本になっているが、分量を考えると値段は手頃と言える。 著者が内容を 自分のウェブサイト で公開しており、実際のところ本を買わなくても内容にアクセスできてしまう。 |
|
Sievers et al., 2011. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539. Link to the paper.Notredame et al., 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 8, 205-217.Pais et al., 2017. Assessing the efficiency of multiple sequence alignment programs. Algorithms Mol Biol 9, 4.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。